 Virus Ecology and Evolution (VEE)
Virus Ecology and Evolution (VEE)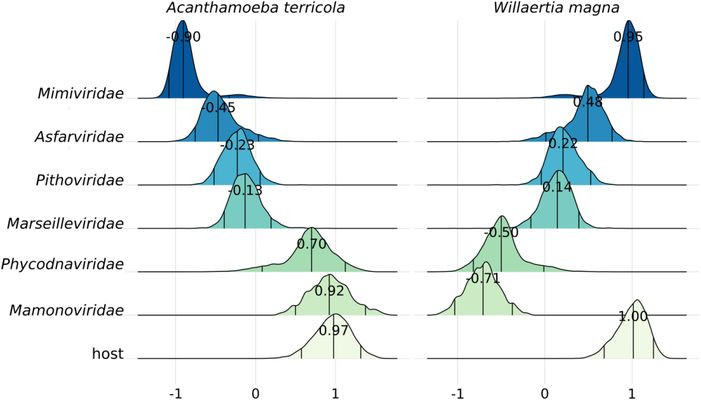
We sequenced high-quality genomes of protists known as hosts of giant viruses (GVs). Matching of codon usage preferences is often used to predict virus-host pairs. Our analyses revealed that in GVs, codon usage alone is a poor predictor of known pairs. Why? well, GVs have complex genomes… They encode few to complete tRNA sets and even genes from the translational machinery (e.g. tRNA–ligases). The host immune system may also play a role, driving viral codon usage away from its own. Moreover, its replication site (nuclear vs. cytoplasmatic) could also play a role. Finally, by analyzing the new amoebal genomes, we discovered viral integrations (potentially from GVs) into some of them, indicating historical infections. Most notably some inserted major capsid proteins seem to potentially encode for intact proteins (work in progress)…
Reference: 10.1101/2024.09.23.614596
*Version of record: 10.1093/gbe/evae271